Temas
Las terapias contra el cáncer fallan cuando las células cancerosas se adaptan al tratamiento debido al aprovechamiento de mecanismo de adaptación está íntimamente relacionado con la capacidad de replicar (duplicación) el ADN en condiciones adversas. El objetivo de nuestro laboratorio es identificar métodos para perturbar de forma potente y selectiva la replicación del ADN de las células cancerosas, evitando así su multiplicación.
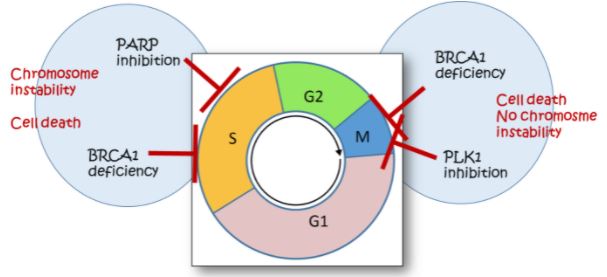
Trabajamos bajo la hipótesis de que la replicación del ADN es un blanco terapéutico contra el cáncer. Cuando el ADN es dañado, por ejemplo, por quimioterapia, las células normales evitan duplicar su ADN, pero las células cancerosas utilizan los templados dañados como molde para copiar ADN. Esto crea una ventana de oportunidad para el desarrollo de tratamientos: si la quimioterapia reduce la calidad de los templados de ADN por debajo de un umbral aceptable, la duplicación del ADN puede detenerse irreversiblemente y al no poder completarse, desencadenar muerte celular. Pero décadas de uso de quimioterapia en seres humanos han revelado que dicho presupuesto no es tan alcanzable como se esperaba inicialmente. Tal limitación es consecuencia de la activación de una red molecular, la respuesta al ADN dañado o DDR (del inglés, DNA damage response), que permite la utilización de templados de ADN dañado como moldes replicativos. Dicha red es esencial para propiciar la sobrevida de células sanas ante estrés metabólico o ambiental; desafortunadamente, las células cancerosas también aprovechan esa DDR para completar la replicación del ADN dañado, sobrevivir y adaptarse a la quimioterapia.
El objetivo principal de nuestro laboratorio es el de dilucidar la contribución de diferentes factores de DDR a la supervivencia de las células cancerosas y a su capacidad de adaptación al tratamiento. Buscamos además interpretar dicho conocimiento en el marco de la identificación de dianas farmacológicamente regulables que sensibilizan selectivamente las células cancerosas a la muerte celular y/o inhiben la adaptación tumoral a la quimioterapia.
Enfoque
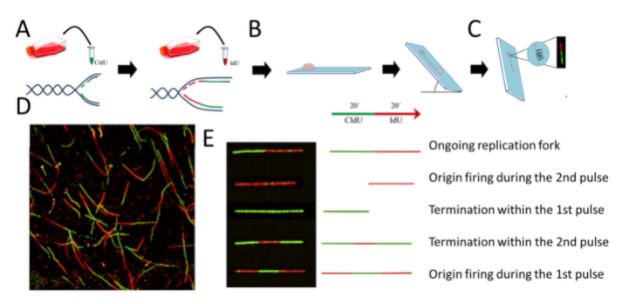
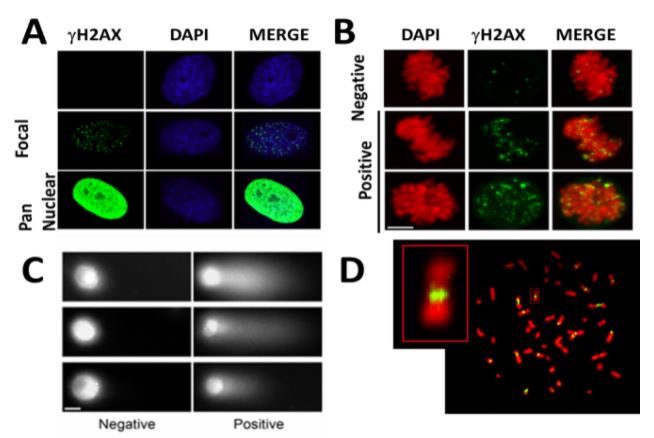
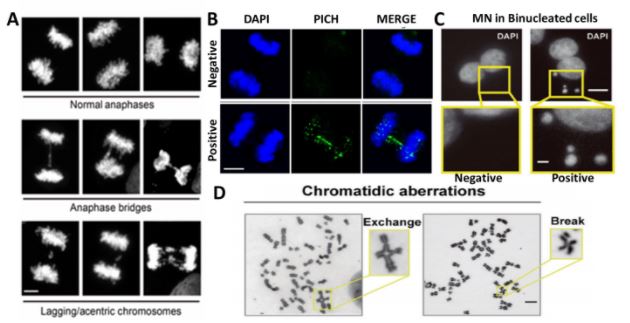
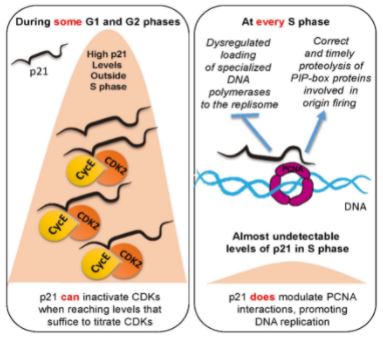
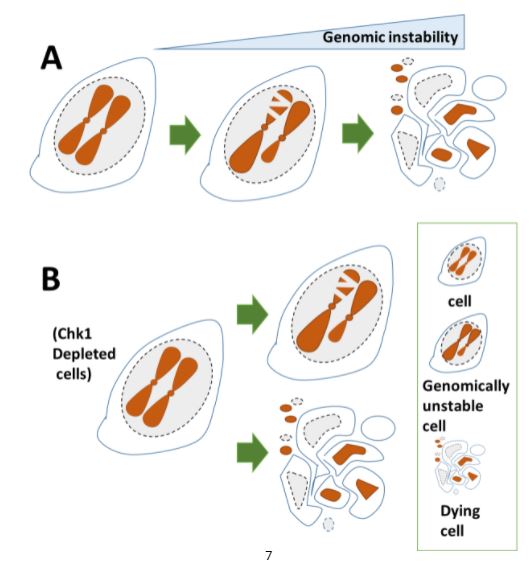
Las herramientas de biología molecular y biología celular de diferentes tipos son fundamentales para nuestra investigación. El sistema modelo que nos permite recapitular la respuesta de las células tumorales al tratamiento son las líneas de células cancerígenas en cultivos in vitro. Aunque simplificadas y artificiales en cuanto a su capacidad de recapitular la complejidad de tejido tumoral, las células cancerosas en cultivo son fáciles de manipular, pudiéndose alterar la expresión de genes específicos con protocolos sencillos. El hacerlo nos permite establecer relaciones causales entre la eliminación/inactivación de un determinado gen y el cambio en una variable funcionalmente relevante. Para revelar variables claves en la red molecular objeto de nuestro estudio, monitoreamos: la replicación del ADN a diferentes niveles, incluida la evaluación de: moléculas individuales de ADN recién sintetizado (Figura 1), diferentes marcadores directos o indirectos de daño del ADN (Figura 2), parámetros de inestabilidad en cromosomas (Figura 3). y muerte celular (Figura 4) entre otros marcadores. Al analizar dichos parámetros, inferimos el potencial efecto terapéutico que pudiese tener la inhibición de un factor celular dado, en cuanto a su capacidad de inducir muerte celular o de prevenir la adaptabilidad de la célula cancerosa al tratamiento.